欧盟体外诊断新法规主要变化情况如何?
(原创2017-10-19CMDE中国器审)
2017年5月5日,欧盟官方期刊(Official Journal of the EuropeanUnion)正式发布了欧盟体外诊断医疗器械法规(IVDR,EU 2017/746)。2017年5月25日,IVDR正式生效,体外诊断医疗器械指令(IVDD,98/79/EC)被体外诊断医疗器械法规取代。IVDR从几个方面对原有IVDD框架进行了修订与完善,如定义与概念、各方职责与义务、风险分类管理、符合性评估流程、公告机构的指定与管理、产品性能评估和性能研究、加强市场监管要求等。IVDR法规共九章113条,并附有15个附录。
一、关于法规过渡期
法规过渡期为5年,共涉及四个时间点(见表1):
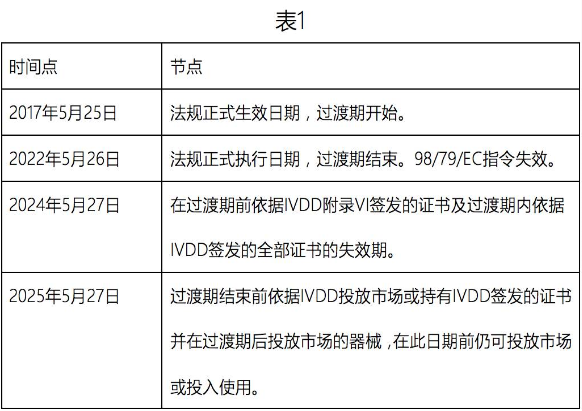
过渡期结束后,所有体外诊断器械必须满足IVDR的要求,即自2022年5月26日起,公告机构依据IVDD发布的任何通知将失效。
在2017年5月25日之前,公告机构根∴据IVDD签发的证书在证书所示期限到期前继续有效,但是根据IVDD附录VI签发的证书应在2024年5月27日之前▓失效。
自2017年5月25日起,公告机构根据IVDD签发的证书在2024年5月27日之前失效。仅具有根据IVDD规定签发的证书的器械可投放市场的前提是卐自IVDR适用之日起,其在设计和预期的目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。
在过渡期结束(2022年5月26日)之前,根据IVDD投放市场的器械及自2022年5月26日起投放市场并具有证书的器械在2025年5月27日之前可继续投放市场或投入使用。
通过IVDD豁免,符合新法规的器械可在2022年5月26日之前投放市场。并可在2022年5月26日前指☆定并通知符合新法规的符合性评估机构。公告机构可在2022年5月26日前,采用其规定的符合性评估流程并按照新法规签发证书。
对于高风险产品(需经D类产品符合性评估流程评估的产品),在已委派必要的医疗器械协调小组(MDCG)、专家小组和欧盟参考实验室前提下,同样可通过IVDD豁免在2022年5月26日之前投放市场。
制造商需在过渡期内更新技术文件和流程以满足新法规更严格的要求。
二、关于公告机构
整体来讲,欧盟新法规对公告机构的管理是以成员国主管机构为主体组织实施,欧盟理事会、欧盟级别的专家理事会参与并监管的模式开展的对公告机构的评估,并在委任后实施持续监管、持续审核的管理。
欧盟新法规对于公告机构的管理更为严格,所有公告机构需要获得欧盟主管当局的认可后,才能依据IVDR进行审核。一般公告机构都会在过渡期内维持IVDD的审核资质。只要公告机构具有IVDD的审核资质,可以申请认证。资质证书的︾有效期是有限的。
三、产品分类的变化
IVDD基于清单分类(list-basedsystem),将产品〗分为List A,ListB和其他。而IVDR基于规则分类(rule-based classification system),将产品按照风险等级分成A类(最低风险),B类,C类和D类(最高风险)。与IVDD的清单分类相比,IVDR的规则分类更为复杂。产品的风险等级是由产品的预期用途和被测量◤的分析物决定的,和产品的命名无关。如果产品适用多个分类,则应遵循最高分类原则。
如D类包含:(1)检测血液、血液成分、细胞、组织或器官、或其任何衍生物是否存在或显露传染性因子,以评估它们是否适用于输血、器官移植或细胞给药;(2)检测是否存在或显露传染性因子,其■会导致危及生命的疾病,并且具有高的或可疑的高传播风险;(3)确定危及生命的疾病的、其监控对于患者管理的过程十分关键的病原体载量;(4)用于血型分型或组织分型中的ABO系统、Rh系统、KELL系统、KIDD系统及DUFFY系统的器ㄨ械。D类均为高风险产品。C类则包括具有较高风险的器械,如用以确保用于输血或移植或细胞给药的血液、血液成分、细胞、组织或器官具有免疫相容性的其他血型分型或组织分型器械,以及用于检测是否存在或显露性传播病原体的♂器械、伴随诊断用器械、新生儿先天性疾病筛查等。A类则相对风险较低,包括适用于相关特定的体外诊断检测流程的一般实验室使用的产品、没有危险特征的附件、缓冲液、洗涤液、一般培养基和组织学染色液等;体外诊断用途专用设备;样品容器。其余未被涵盖的器械归为B类,不具有定量或定性赋值的质控品类器械归类为B类。
产品的¤分类由制造商负责。如果公告机构对产品分类有疑义,公告机构可向主管当局进行咨询。如果公告机@构的主管当局和制造商的主管当局咨询的意见仍不一致,医疗器械协调组(MDCG)将参与边界产品分类的决策。
四、关于符合性评估
基于分类原则的产品风险分类,体现在产品分级监管々中。在器械投放市场前,不同类别的产品依照IVDR附录IX到XI的符合性评估流程,进行符合性评估。D类器械的符合性评◥估有两条途径:
途径一:基于质量管理体系和技术文件评估的符合性评估
包括相关器械全生命周期的质♀量管理体系评估、监管评估、技术文件评估以及上市后主管机构监督要求。制造商需向公告机构提出申请,评估自己的质量管理体系,进行监管评估以确保制造商充分履行批准后的质量管□理体系所规定的义务,并申请进行相关器械技术文件评估,在最后一个器械上市后不迟于10年期限内,制造商或其授权代表应受主管机构监管。
途径二:基于型式检验的符合性评估和基于生产质量保证的符合性评估
EU型式检←查是公告机构确定和证明器械(包括其技术文件和相关生命周期过程以及所涵盖的相应代表性器械样品)符合法规相关规∮定要求的程序。制造商应向公告机构提出申请并提交申报文件,由公告机构进行评估,包括技术文件、数据的评估及检验试验过程。基于生№产质量保证的符合性评估包括公告机构对实施批准的医疗器械生产质量管理体系的评估、体系监管评估、所生产器械的验证。
上述两项评估程序均要求制造商或其授权代表在最后一个器械↓上市至少10年内,保管相应文件。
C类器械的符合性评估▲较D类有所简化,主要包括质量管理体系评估、体系监管评估和上市后主管机构监督要求,并且每个同类器械组应评估至少一个典型器械的技术文件。
B类器械的符合性评』估与C类相似并更为简化,主要包括质量管理体系评估和上市后主管机构监督要求,并且每个器械类别应评估至少一个典型器械的技术文件。
另外法规中⌒ 还对自测用器械、伴随诊断器械等符合性评估规定了特殊要求。
公告机构在符合性评估后颁发证书,证书有效期为▓其列明的期限,不超过五年。经制造商申请,证书的有效期可以延长,但每次延期不超过五年,同时需按照适用的符合性评估流程重新评估。
A类器械制造∮商在拟定符合IVDR附录II(技术文件)和附录III(关于上市后监管的技术文件)规定的技术文件后,通过签发EC符合性声明的形式,声明其产品的符合性。基于现有IVDD指令,只有约不到20%的≡体外诊断产品需要公告机构参与,但在IVDR法规下,预计将有约超过80%的体〓外诊断产品需要公告机构参与。
五、关于上市后监管、警戒和市场监管
作为制造商质量管理体系的组成部分,制造商应采用与风险等级相称并且使用与该器械类型相适应的方式来计划、建立、记录、实施、维护和更新上市后的监◥管体系。不同分类的产品上市后监管报告要求不同,A类和B类器械的制造商应编制一份上市后监管报告,总结根据上市后监管计划收集的上市后监管数据的分析结果和结论,以及采取任何预防和整改措施的理由和描述。应在必要时对报告▂进行更新,并应按照相应要求提供给公告机构和主管机构。
C类和D类器械的制造商应编制定期安全性更新报告(PSUR),总结根据上市后监管计划收集的上市后监管数据的分析结果和结论,以及采取任何预防和整改措施的理由和描述。C类和D类器械的制造商应至少每年对PSUR进行更新。PSUR应属于法规规定技术文件中的一部分。
D类器械的制造∩商应通过电子系统的方式,并向参与符合性评估的公告机构提交PSUR。公告机构应审查该报告,并将其评估添加到该电子系统中,评估中应包括采取任何措施的细节。PSUR和公告机构的评估应通过电子系统提供@ 给主管机构。
C类器械制造商应向参与符合性评估的公告机构提交PSUR,并应主管机构要求向其提供报告。
制造商应向主管机构报告严重事件和现场安全整改措施。并对警戒数据进行分析。主管机构应主动参与相关程序,协调评估。
主管机构负有市场监督责◆任,对器械的符合性特性和性能执行适当的检查,包括酌情审查文件以及基于适当样品的物理或实验室检查。必要时可在欧盟层面评估国家措施是否合理。IVDR从各个层面对体外诊断医疗器械的监管提出了︻要求,内容丰富,技术要求如第VI章,对临床证据、性能评估和性能研究的规定,政策层面如对欧盟成员国之间的协调与合作的要求,对公告机构的管理、对电子系统的要求,对器械唯一标识系统(UDI系统)等方面的√要求,更为详细具体。整体法规趋于更为严格的管理模式,如对制造商的要求、对主管机构职责的明确、对公告机构的管理以及对产品基于风险分类的上市前和上市后管理等。具体IVDR评审要求的变化将取决于欧盟委员会的指导文件,这些具体的指导文件尚待发布。
审评六部吕允凤供稿
2017-11-02 15:11