ICHΒΎΕΰΫχ≥ΧΘ§”Ύ2008Ρξ7‘¬25»’ΖΔ≤Φ
…σΤάΕΰ≤Ω ΜΤΖΦΜΣΘ®“κΘ©
7Θ°ΧΫΥς–‘ΝΌ¥≤ ‘―ι
‘Ύ“Μ–©«ιΩωœ¬Θ§‘γΤΎΜώΒΟ»ΥΧε”Π”Ο ΐΨί±Μ»œΈΣ”–άϊ”ΎΩΦ≤λ»ΥΧε…ζάμΓΫ―ß/“©άμ―ßΓΔΚρ―Γ“©ΈοΧΊ’ςΚΆ”κΦ≤®v≤ΓœύΙΊΒΡ÷ΈΝΤΑ–ΒψΓτΒΡ–≈œΔΓΘΉν–¬ΒΡΘ®StreamlinedΘ©‘γΤΎΧΫΥς–‘ΖΫΖ®Ρή¥οΒΫΗΟΡΩ±ξΓΘ±Ψ÷ΗΒΦ‘≠‘ρ÷–ΒΡΧΫΥς–‘ ‘―ι «÷ΗΡ«–©Ρβ‘ΎΔώΤΎ ‘―ι«ΑΫχ––ΓΔΑϋΚ§”–œόΒΡ»ΥΧ屩¬ΕΓΔΖ«÷ΈΝΤΓϊΚΆ’οΕœΡΩΒΡΘ§“‘ΦΑΖ«“β‘ΎΦλ≤βΉν¥σΡΆ ήΝΩΒΡ ‘―ιΓΘΥϋΟ«®L”Ο”Ύ―–ΨΩΕύ÷÷≤Έ ΐΘ§»γ“©¥ζΕ·ΝΠ―ßΓΔ“©–ß―ßΚΆΤδΥϊ…ζΈο÷Η±ξΘ§Αϋά®PET ήΧεΫαΚœΚΆ÷ΟΜΜΓΘ
‘Ύ’β–©«ιΩωΘά œ¬Θ§ “ΥΒΡΖ«ΝΌ¥≤÷ß≥÷ ΐΠΊΓΓΨίΒΡΝΩ»ΓΨω”ΎΗυΨίΉν¥σΝΌ¥≤ Ι”ΟΦΝΝΩΚΆ”Ο“©®wΤΎœόΕχά¥ΒΡΡβΕ®ΝΌ¥≤±©¬ΕΒΡ≥ΧΓΩΕ»ΓΘΉςΈΣάΐΉ”Θ§œ¬ΟφΘ®ΚΆ‘Ύ±μ3÷–ΗϋœξœΗΟη ωΘ©Οη ωΝΥ5÷÷≤ΜΆ§ΒΡΧΫΥς–‘ΝΌ¥≤ ‘―ιΖΫΖ®Θ§“‘ΦΑ’β–©ΧΊ βΖΫΖ®÷–“Σ«σΒΡ®ηΖ«ΝΌ¥≤ ‘―ιΫχ≥ΧΓΘΒΪ «Θ§‘Ύ±Ψ÷ΗΒΦ‘≠‘ρ÷–Έ¥Οη ωΒΫΒΡΤδΥϊΖΫΖ®“≤Ω…≤…”ΟΓΘ’β–©”Π”κΓτœύ”ΠΒΡΙήάμΒ±Ψ÷Ϋχ––Χ÷¬έ≤ΔΜώΒΟ»œΆ§ΓΘ’β–©ΖΫΖ®ΒΡ Ι”Ο÷Φ‘ΎΦθ…Ό“©ΈοΩΣΖΔ÷–Υυ Ι”ΟΒΡΕ·ΈοΉή®¨ ΐΓΘ
±μ3÷–Αϋά®ΝΥΕ‘”ΎΟΩ÷÷ΖΫΖ®Κœ ΒΡΤπ ΦΦΝΝΩΚΆ÷’Γβ÷ΙΦΝΝΩΓΘ‘ΎΗς÷÷«ιΩωœ¬Θ§≤…”ΟΧεΡΎΚΆ/ΜρΧεΆβΡΘ–ΆΟη ω“©¥ζΕ·ΝΠ―ßΚΆ“©άμ―ßΧΊ’ς «÷Ί“ΣΒΡΘ§≤Δ”Π”ΟΤδά¥÷ß≥÷»ΥΧεΦΝΝΩ―Γ‘ώΓΘ
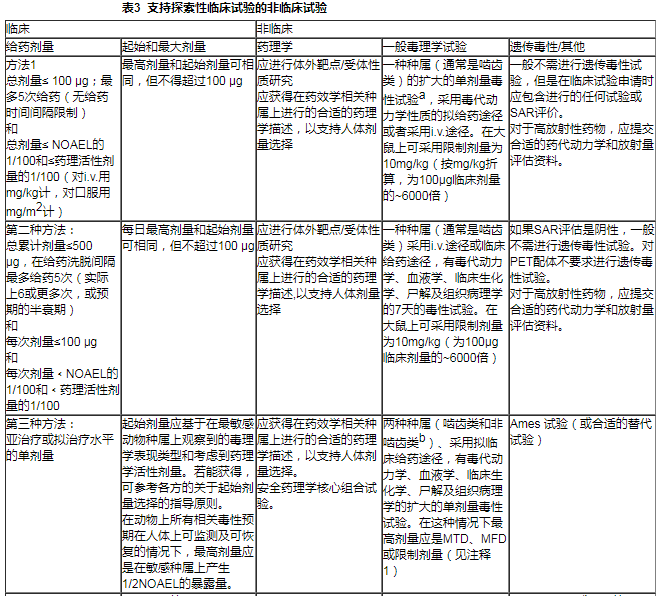
7Θ°1ΈΔΦΝΝΩΓ‘ ‘―ι
œ¬ΟφΟη ωΠγΝΥΝΫ÷÷≤ΜΆ§ΒΡΈΔΦΝΝΩ ‘―ιΖΫΖ®ΓΘ
ΒΎ“Μ÷÷ΖΫΖ® «Θ§ ή ‘’ΏΉήΦΝΝΩ≤Μ≥§Ιΐ100 ΠΧgΘ§‘ΎΟΩΗω ή ‘’Ώ…œΉνΕύΩ…Ζ÷5¥ΈΗχ“©ΓΘPET ‘―ι”–÷ζ”Ύ―–ΨΩΑ– ήΧεΫαΚœΜρΉι÷·Ζ÷≤ΦΓΘΒΎΕΰΗω”ΟΆΨ «”–ΜρΈόΆ§ΈΜΥΊ±ξΦ«Έο Ι”Οœ¬ΤάΙά“©¥ζΕ·ΝΠ―ßΓΘ’β–©”ΟΆΨΩ…”…‘Ύ“Μ÷÷÷÷ τ…œΘ®Ά®≥Θ «®LΡω≥ίάύΘ©Ϋχ––ΒΡΓΔ≤…”ΟΝΌΓω¥≤Ηχ“©ΆΨΨΕΒΡά©¥σΒΡΒΞΦΝΝΩΕΨ–‘ ‘―ιΘ§“‘ΦΑΚœ®è ΒΡ“©άμ―ßΧΊ’ςΟη ωά¥÷ß≥÷ΓΘ
ΒΎΕΰ÷÷ΓχΈΔΦΝΝΩΖΫΖ® «Θ§ΑϋΚ§–Γ”Ύ 5¥ΈΒΡΗχ“©Θ§ΤδΉνΗΏΗχ“©ΝΩΈΣ100ΠΧg/¥ΈΘ®ΟΩ ή ‘’ΏΉήΦΝΝΩ500ΠΧgΘ©ΓΘΆ§…œ ωΒΎ“Μ÷÷ΖΫΖ®Θ§±ΨΖΫΖ®”–άύΥΤΒΡ”ΟΆΨΘ§ΒΪ «≤…”ΟΗϋ…ΌΒΡΜν–‘PET≈δΧεΓΘΗΟΖΫΖ®Ω…”…‘ΎΓΤΓΓ“Μ÷÷÷÷ τ…œΘ®Ά®≥Θ «Ρω≥ίάύΘ©Ϋχ––ΒΡΓΔ≤…”ΟΝΌ¥≤Ηχ“©ΆΨΨΕΒΡ7ΧλΕΨ®y–‘ ‘―ιΓΔ“‘ΦΑΗΟΈ¥±ξΦ«Μ·ΚœΈοΒΡ“≈¥ΪΕΨΓ‘–‘«±ΝΠΒΡSARΘ®Α≤»ΪΓωΖ÷Έω±®ΗφΘ©ΤάΙά”κΚœ ΒΡ“©άμ―ßΧΊ’ςΟη ωά¥÷ß≥÷ΓΘ
‘ΎΡ≥–©«ιΩωœ¬Θ§Ε‘”Ύ“ΜΗωΡβΩΎΖΰΗχ“©ΚΆ“―”–ΩΎΖΰΗχ“©Ζ«ΝΌ¥≤ΕΨάμ ‘―ιΒΡ®à≤ζΤΖΘ§≤…”Οi.v.ΆΨΨΕΫχ––ΝΌ¥≤ΈΔΦΝΝΩ ‘―ι «Κœ ΒΡΓΘ‘ΎΗΟ÷÷«ιΩωΓχœ¬Θ§ i.v.ΈΔΦΝΝΩ”…“―”–ΒΡΓςΩΎΖΰΗχ“©÷ΊΗ¥ΦΝΝΩΕΨ–‘ ‘―ιΘ®Τδ“―¥οΓΙΒΫ≥δΖ÷ΒΡ±©¬ΕΖΕΈßΘ©ά¥÷ß≥÷ΓΘ”…”ΎΗΟΗχ“©ΦΝΝΩ®ηΦΪΒΆΘ®Φ¥ΉνΗΏΦΝΓ©ΝΩ100 ΠΧgΘ©Θ§‘Ύ’β÷÷«ιΩωœ¬≤Μ“Σ«σΩΦ≤λi.v.Ηχ“©ΒΡΨ÷≤ΩΡΆ ή–‘ΓΘ
7Θ°2ΦΝΝΩ÷Ν―«÷ΈΝΤΦΝΝΩΜρΡβΕ®÷ΈΝΤΦΝΝΩΖΕΈßΒΡΒΞΦΝΝΩ ‘―ι
ΒΎΓΦ»ΐ÷÷ΖΫΖ® «Θ§ ή ‘’ΏΗχ”ηΦΝΝΩ÷Ν―«÷Έ®KΝΤΥ°ΤΫΘ®“©άμ―ßΘ©Μρ÷ΈΝΤΖΕΈßΒΡΒΞΦΝΝΩΘ®Φϊ±μ3Θ©ΓΘΉν¥σΩ…‘ –μΦΝΝΩά¥‘¥”ΎΖ«ΠΈΓΓΝΌ¥≤ ΐΨίΓΘΥϋΫχ“Μ≤Ϋ ήΒΫ»ΥΓ―ΧεΒΞΦΝΝΩ ‘―ι÷–ΥυΜώΒΟΒΡΝΌ¥≤–≈œΔΒΡœό÷ΤΓΘ≤…”Ο®}Ζ«Ζ≈…δ±ξΦ«ΒΡ“©ΈοΓΔ‘ΎΜρΫ”Ϋϋ‘ΛΤΎΒΡ“©–ß―ßΜν–‘ΦΝΝΩ ±Θ§ΗΟΖΫΖ®Ω…»ΖΕ®“©¥ζΕ·ΝΠ―ß≤Έ ΐΘ®»γt1/2 Μρ…ζΈοάϊ”ΟΕ»Θ©ΓΘΗΟΖΫΖ®”…“Μ÷÷Ρω≥ίάύΚΆ“Μ÷÷Ζ«Ρω≥ίάύΝΫ÷÷Ε·ΈοΒΡΓβά©¥σΒΡΒΞΦΝΝΩΕΨ–‘ ‘―ιΓΔ“ΜœνΓΨ“≈¥ΪΕΨ–‘ΤάΙά ‘―ιΘ®Ames ‘―ιΘ©ΓΔΚœ ΒΡ“©άμ―ßΧΊ’ςΟη ωΚΆΓΗΑ≤»Ϊ“©άμ―ßΚΥ®L–Ρ ‘―ιά¥÷ß≥÷ΓΘ
7Θ°3ΕύΦΝΝΩΓτ ‘―ι
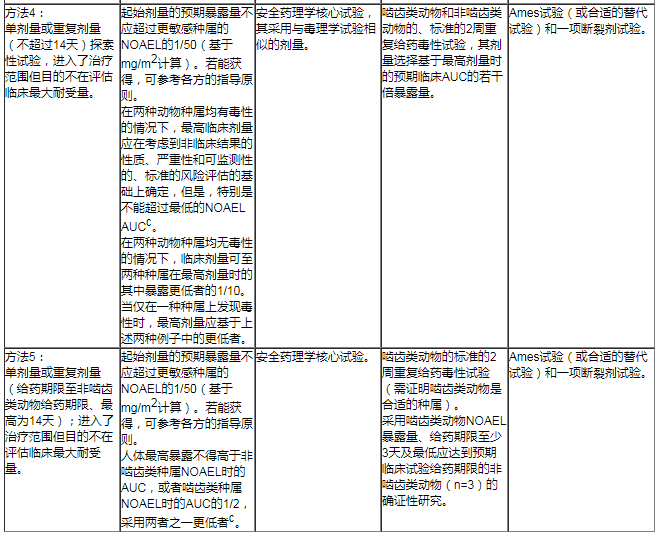
ΒΎΥΡ÷÷ΚΆΒΎΈε÷÷ΖΫΖ®Θ§ «”Ο”Ύ»ΖΕ®»ΥΧεΒΡ“©¥ζΕ·ΝΠ―ßΚΆ“©–ß―ß“‘ΦΑΤδΡΩΒΡΖ«÷ß≥÷Ήν¥σΡΆ ήΝΌ¥≤ΦΝΝΩΒΡ»ΖΕ®ΓΔ≤Μ≥§Ιΐ14ΧλΗχ“©ΒΡ ‘Γπ―ιΓΘ
ΒΎΥΡ÷÷ΖΫΖ®Αϋά®Ρω≥ίάύΚΆΖ«Ρω≥ίάύΝΫ÷÷Ε·ΈοΒΡ2÷ή÷ΊΗ¥Ηχ“©ΕΨ–‘ ‘―ιΘ®ΤδΦΝΝΩ―Γ‘ώΜυ”ΎΉν¥σΝΌ¥≤ΦΝΓΐΝΩ ±ΒΡ‘ΛΤΎAUCΒΡΘά »τΗ…±Ε±©¬ΕΝΩΘ©ΓΘ»τ’β–© ‘―ι‘ΎΘάΝΫ÷÷÷÷ τ…œ≥δΖ÷Οη ωΝΥΗΟΜ·ΚœΈοΕΨ–‘Θ§ΧΫΥς–‘ΝΌ¥≤ ‘―ιΒΡΉνΗΏΦΝΝΩ”ΠΗυΨί±ξΉΦΒΡΑ≤»Ϊ–‘/Ζγœ’–‘ΤάΙάΘ®ΩΦ¬«ΒΫΖ«ΝΌ¥≤ΫαΙϊΒΡ–‘÷ Γ”ΓΔ―œ÷Ί≥ΧΕ»ΓΔΦύ≤β÷Η±ξΘ©ά¥»ΖΕ®ΓΘ‘ΎΝΫ÷÷Ε·Έο÷÷ τ»±ΖΠΕΨ–‘ΒΡ«ιΩωœ¬Θ§ΝΌ¥≤ΠΤΦΝΝΩ÷ΝΝΫ÷÷÷÷ τ‘ΎΉνΗΏΦΝΝΩ ±ΒΡΓ©Τδ÷–±©¬ΕΝΩΗϋΒΆ’ΏΒΡ1/10»œ±ΜΈΣ «Κœ ΒΡΓΘΒ±Ϋω‘Ύ“Μ÷÷÷÷ τ…œΖΔœ÷ΕΨ–‘ ±Θ§ΉνΗΏΝΌ¥≤ΦΝΝΩ‘ρ”ΠΜυ”Ύ…œ ωΝΫ÷÷άΐΉ”÷–ΒΡΗϋΒΆ’ΏΓΘ
ΒΎΈε÷÷ΖΫΖ®Αϋά®‘Ύ“Μ÷÷Ρω≥ίάύΕ·Έο…œΫχ––ΒΡΓΔΗχ“©ΦΝΝΩ¥οΒΫΉνΗΏΡΆ ήΝΩΒΡ2÷ήΕΨ–‘ ‘―ιΘ§“‘ΦΑΡΩΒΡ‘Ύ”Ύ―Α’“÷Λ ΒΡω≥ίάύΕ·ΈοΒΡNOAELΕ‘Ζ«Ρω≥ίάύΕ·Έο“≤ «ΈόΕΨ®ç–‘ΦΝΝΩΒΡ»Ζ÷Λ–‘Ζ«ΡωάύΕ·Έο ‘―ιΓΘ»Ζ÷Λ–‘Ζ«Ρω≥ίάύΕ·Έο ‘―ιΘ§ΑϋΚ§ΝΥ÷ΊΗ¥Ηχ”ηΡω≥ίάύΕ·Έο≤ζ…ζΓβNOAEL±©¬ΕΝΩΒΡΦΝΝΩΘ®≥Θ≥ΘΗυΨίΧε±μΟφΜΐΜρ ΒΦ ΜρΡΘΡβΒΡ±©¬Εά¥ΙάΥψΘ©ΓΘΖ«Ρω≥ίάύΕ·Έο ‘―ιΗχ“©ΤΎœό÷Ν…ΌΈΣ3ΧλΘ§«“÷Ν…Ό”Π”κΝΌ¥≤ ‘―ιΡβΗχ“© ±ΦδœύΆ§ΓΘ
Μρ’ΏΘ§Ω…Ϋχ––Ζ«Ρω≥ίάύΒΡΦΝ®uΝΩΒί‘ω ‘―ιΘ§ΗΟΓω ‘―ι÷–‘ΎΗχ“©Ϋα χ ±Ε‘Ε·ΈοΗχ”ηΝΥΡω≥ίάύΕ·Έο‘ΛΤΎNOAELΒΡ±©¬ΕΝΩΓ‘÷Ν…Ό3ΧλΓΘ”Π÷ΛΟςΖ«Ρω≥ίάύΕ·Έο±»Ρω≥ίάύΕ·ΈοΗϋΟτΗ–Θ§ΝΌΓό¥≤Ηχ“©”Π―”≥Ό÷Ν‘ΎΗΟ÷÷ τ…œΫχ––ΝΥΗϋΫχ“Μ≤ΫΒΡΖ«ΝΌ¥≤ ‘―ιΘ®Ά®≥Θ «±ξΉΦΒΡΕΨ–‘ ‘―ιΘ©Κσ≤≈Ϋχ––ΓΘ
ΝΫ÷÷ΖΫΖ®Ψυ–η“ΣΫχ––“≈¥ΪΕΨ–‘ΤάΙάΘ®Ames ‘―ιΚΆ“ΜœνΕœΝ―ΦΝ ‘―ιΘ©ΓΔΑ≤»Ϊ“©άμ―ßΚΥ–Ρ ‘―ιΘ®ΤδΩ…ΒΞΕάΫχΓυ–– ‘―ιΘ§ΜρΑϋΚ§‘Ύ“Μœν÷ΊΗ¥Ηχ“©ΕΨ–‘ ‘―ι÷–Θ©ΓΘ
aΘ°Ά®≥ΘΘ§ΈΣΓ‘÷ß≥÷ΝΌ¥≤Ηχ“©ΦΝΝΩΘ§ά©¥σΒΡΒΞΦΝΝΩ ‘―ι…ηΦΤ”Π‘ΎΒΎ2Χλ ±ΟΩΉιΟΩ®΄–‘±π10÷ΜΡω≥ίάύΕ·ΈοΓΔΒΎ14ΧλΟΩΉιΟΩ–‘±π50÷ΜΡω≥ίΓΤάύΕ·ΈοΒΡ―Σ“Κ―ßΓΔΝΌ¥≤…ζΜ·―ßΓΔ §ΦλΚΆΉι÷·≤Γάμ―ßΦλ≤ιΫχ––Άξ’ϊΓ«ΤάΦέΓΘ
bΘ°Ε‘”ΎΡω≥ίάύΕ·ΈοΘ§Φϊ…œΓΘΕ‘”ΎΖ«ΓΙΡω≥ίάύΕ·ΈοΘ§2Χλ ±ΟΩΓΖΉιΟΩ–‘±π3÷ΜΘ§14Χλ ±‘ρΫωΕ‘ΗΏΦΝΝΩΟΩΉιΟΩ–‘±π2÷ΜΫχ––Φλ≤ιΓΘ
cΘ°‘Ύ»±ΖΠΝΌ¥≤≤ΜΝΦΖ¥”ΠΉς”ΟΒΡ«ιΩωœ¬Θ§»γΙϊΕΨάμ―ßΫαΙϊ‘Ύ»ΥΧε…œ‘ΛΤΎΩ…Φύ≤βΓΔΩ…Μ÷Η¥“‘ΦΑ―œ÷Ί–‘ΫœΒΆΘ§ΗΏ”ΎAUCΒΡΦΝΝΩ «ΓρΚœ ΒΡΓΘ
8Θ°Ψ÷≤ΩΡΆ ή–‘ ‘―ι©IΓΓ
ΫΪ≤…”ΟΡβΝΌ¥≤Ηχ“©ΆΨΨΕΒΡΨ÷≤ΩΓΐΕΨ–‘ΤάΦέΉςΈΣ≥ΘΙφΕΨ–‘ ‘―ιΒΡ“Μ≤ΩΖ÷®KΗϋΈΣΚœ ΘΜ≤ΜΆΤΦωΫχ––ΒΞΕάΒΡ ‘―ιΓΘ
ΈΣ÷ß≥÷≤…”ΟΖ«÷ΈΝΤΗχ“©ΆΨΨΕΒΡ”–œό÷ΤΒΡ»ΥΧεΗχ“©Θ®»γΒΞ¥Έi.v.Ηχ“©“‘÷ζ”Ύ»ΖΕ®ΩΎΖΰ“©ΈοΒΡΨχΕ‘…ζΈοάϊ”ΟΕ»Θ©Θ§‘ΎΒΞ“Μ÷÷ τ…œΫχ––“ΜΗωΨ÷≤ΩΡΆ ή–‘ ‘―ι±Μ»œΈΣ «Κœ ΒΡΓΘΒ±“―”–ΒΡΕΨάμ ‘―ιΉιΚœΑϋΚ§ΝΥΓοΖ«÷ΈΝΤΗχ“©ΆΨΨΕΒΡ‘ΛΤΎ»Ϊ…챩¬ΕΘ®AUC ΚΆCmax) ±Θ§Ψ÷≤ΩΡΆ ή–‘ ‘―ιΒΡ÷’Βψ÷Η±ξΩ…œό”ΎΝΌ¥≤÷ΔΉ¥ΓΔΉΔ…δ≤ΩΈΜ»β―έΦλ≤ιΚΆΨΒœ¬Φλ≤ιΓΘ
Ε‘”Ύ”…ΓΩΩΎΖΰΕΨάμ ‘―ιΉιΚœΘ®ΦϊΒΎ7ΫΎΘ©÷ß≥÷ΒΡ“ΜΗωi.v.ΈΔΦΝΝΩ ‘―ιΘ§Έό–ηΫχ––Ψ÷≤ΩΡΆ ή–‘ΤάΦέΘ§≥ΐΖ«Τδ÷– Ι”ΟΝΥ–¬»ήΟΫΓΘ
Ε‘”ΎΉΔ…δ”Ο“©Θ§Β±Ω…–– ±Θ§”Π‘Ύ¥σ―υ±ΨΜΦ’Ώ±©¬ΕΘ®»γΔσΤΎ®ηΝΌ¥≤ ‘―ιΘ©«ΑΕ‘Ζ«ΓΰΡΩΒΡΘ®unintendedΘ©ΉΔ…δ≤ΩΈΜΫχ––Ψ÷≤ΩΡΆ ή–‘ΤάΙάΓΘ‘Ύ≤ΜΆ§ΒΡΗςΖΫ’β–© ‘―ιΖΫΖ®”–Υυ≤ΜΆ§ΓΘΟάΙζ≤Μ“Σ«σ’β–© ‘―ιΓΘ»’±Ψ“Σ«σΗυΨίΨΏΓΚΧε«ιΩω”Ο”Ύi.v.“©ΈοΚΆΤδΥϊΉΔ…δ”Ο“©ΈοΓΘ≈ΖΟΥΕ‘”ΎΥυ”–ΒΡΉΔ…δ”Ο“©ΈοΨυ“Σ«σ’β–© ‘―ιΓΘ
9Θ°“≈¥ΪΕΨ–‘ ‘―ι
ΒΎ7ΫΎΧ÷¬έΝΥ÷ß≥÷ΧΫΥς–‘ΝΌ¥≤ ‘―ιΒΡΆΤΦωΒΡ“≈Γϋ¥ΪΕΨ–‘ ‘―ιΖΫΖ®ΓΘ
Μυ“ρΆΜ±δ ‘―ιΆ®≥Θ±Μ»œΈΣΉψ“‘÷ß≥÷Υυ”–ΓΰΒΡΒΞΦΝΝΩΝΌ¥≤ ‘―ιΓΘ÷ß≥÷ΕύΦΝΓΰΝΩΝΌ¥≤ ‘―ιΒΡΝΫ÷÷ ‘―ιΉιΚœΘ®―Γ‘ώ1ΚΆ―Γ‘ώ2Θ©Θ§‘ΎICH S2(R1)ΈΡΦΰΘ®≤ΈΩΦ8Θ©÷–Ϋχ––ΝΥΟη ωΓΘ»τ≤…”Ο―Γ‘ώ2Θ§”Π‘ΎΕύΦΝΝΩ ‘―ιΒΡ»ΥΧε Ή¥Έ”Π”Ο«ΑΆξ≥…ΓΘ»τ≤…”Ο―Γ‘ώ1Θ§Τδ÷–ΒΡΧεΆβ ‘―ι”Π‘Ύ Ή¥ΈΕύΦΝΝΩ»ΥΧε ‘―ι«ΑΆξ≥…Θ§―Γ‘ώ1ΒΡΧεΡΎ ‘―ιΓθ‘ρ”Π‘ΎΔρΤΎ ‘Γύ―ι«ΑΆξ≥…ΓΘ
»τ≥ωΓ‘œ÷―τ–‘ ‘―ιΫαΙϊΘ§”ΠΫχ––ΓόΤάΦέ“‘ΦΑΩ…Ρή–ηΫχ––Ϋχ“Μ≤Ϋ ‘―ιΘ®≤ΈΩΦ8Θ©Θ§“‘»ΖΕ® «ΖώΜΙ ΚœΕ‘»ΥΧεΦΧ–χ®çΗχ“©ΓΘ
10Θ°÷¬Α©Γΰ–‘ ‘―ι
»γΙϊΗυΨίΝΌ¥≤ ”Π÷ΔΆΤΦωΫχ––÷¬Α©–‘ ‘―ιΘ§”Π‘Ύ…œ –…ξ«κ ±Άξ≥…ΓΘ÷Μ”–‘Ύ”–Οςœ‘“ΐΤπ÷¬Α©–‘Ζγœ’ΒΘ”«‘≠“ρΒΡ«ιΩω®àœ¬Θ§”ΠΧαΫΜΗΟœν ‘―ιΫαΙϊ“‘÷ß≥÷ΝΌ¥≤ ‘―ιΓΘ
ICHΈΡΦΰ÷–Χ÷¬έΝΥ÷¬Α©–‘ ‘―ιœύΓνΙΊΒΡ«ιΩωΘ®≤ΈΩΦ9Θ©ΓΘΕ‘”ΎΡβ÷ΈΝΤ≥…»ΥΜρ©•–ΓΕυΒΡΡ≥–©―œ÷ΊΦ≤≤ΓΒΡ“©ΈοΘ§»τΆΤΓωΦωΫχ––Θ§Τδ÷¬Α©–‘ ‘―ιΩ…‘Ύ“©Έο…œ –ΚσΆξ≥…ΓΘ
11Θ°…ζ÷≥ΕΨΛΉ–‘ ‘―ι
”ΠΧαΙ©”κ”Ο“©»Υ»ΚœύΙΊΒΡ…ζ÷≥ΕΨ–‘ ‘―ιΘ®≤ΈΩΦ9ΓΔ10Θ©ΓΘ
11.1 Ρ––‘
“ρΈΣ÷ΊΗ¥Ηχ“©ΕΨ–‘ ‘―ιΜαΤάΦέ“©ΈοΕ‘–έ–‘…ζ÷≥ΤςΙΌΒΡ”ΑœλΘ®ΉΔ Ά2Θ©Θ§Υυ“‘‘Ύ–έ–‘…ζ”ΐΝΠ ‘―ιΆξ≥…«ΑΘ§Ρ––‘ ή ‘’ΏΩ…“‘»κΓυ―ΓΔώΓΔΔρΤΎΝΌ¥≤ ‘―ιΓΘ
–έ–‘…ζ”ΐΝΠ ‘―ι”Π‘Ύ¥σΙφΡΘΓυΜρ≥ΛΤΎΗχ“©ΒΡΝΌ¥≤ ‘―ιΘ®»γΔσΤΎ ‘―ιΘ©ΩΣ Φ«ΑΆξ≥…Θ®≤ΈΩΦ8Θ§9Θ©ΓΘ
11.2 Έό…ζ”ΐΩ…ΡήΒΡΗΨ®v≈°
»γΙϊ“―Άξ≥…œύΙΊΒΡ÷ΊΗ¥Ηχ“©ΕΨ–‘ ‘―ιΘ®Τδ÷–Αϋά®Ε‘¥Τ–‘…ζ÷≥ΤςΙΌΒΡΤάΦέΘ©Θ§Έό…ζ”ΐΩ…ΡήΗΨ≈°Θ®Φ¥Ψχ”ΐΜρΨχΨ≠ΚσΓμΗΨ≈°Θ©Ω…“‘‘Ύ»±…ΌΕ·ΈοΓ–…ζ÷≥ΕΨ–‘ ‘―ιΒΡ«ιΩωœ¬»κ―ΓΝΌ¥≤ ‘―ιΓΘΨχΨ≠ΚσΕ®“εΈΣ‘ΎΈόΧφ¥ζ“©Έο‘≠“ρΒΡ«ιΩωœ¬ΆΘΨ≠12Ηω‘¬ΓΘ
11.3 ”–Ω…Ρή…ζ”ΐΒΡΗΨ≈°
Ε‘”Ύ”–Ω…Ρή…ζ”ΐΒΡΗΨ≈°(WOCBP)Θ§‘Ύ…–≤Μ«ε≥ΰ«±‘ΎΒΡΖγœ’/–ß“φ ±Θ§”ΠΗΏΕ»÷Ί ”Ε‘≈ΏΧΞΜρΧΞΕυΒΡΖ«‘ΛΤΎΒΡ“©Έο±©¬ΕΓΘΙΊ”ΎΫχ––…ζ÷≥ΕΨ–‘ ‘―ιΒΡ ±Φδ“‘÷ß≥÷”–…ζ”ΐΩ…ΡήΗΨ≈°»κ―ΓΝΌ¥≤ ‘―ιΘ§ΡΩ«ΑICHΗςΖΫΒΡ“Σ«σΓράύΥΤΓΘ
Β±ΝΌ¥≤ ‘―ι÷–Ρ…»κWOCBP ±Θ§±Ί–κ ΙΕ‘≈ΏΧΞΜρΧΞΕυΒΡΖγœ’ΫΒ÷ΝΉνΒΆΓΘΈΣ¥οΒΫΗΟΡΩ±ξ”–ΚήΕύΖΫΖ®ΓΘ“Μ÷÷ΖΫΖ® «Ϋχ––…ζ÷≥ΕΨ–‘ ‘―ι“‘ΝΥΫβ“©Έο±Ψ…μΙΧ”–ΒΡΖγœ’“‘ΦΑ‘Ύ«±‘Ύ±©¬ΕΙΐ≥Χ÷–≤…”ΟΚœ ΒΡ‘ΛΖά¥κ ©ΓΘΒΎΕΰ÷÷ΖΫΖ® «ΝΌ¥≤ ‘―ιΙΐ≥Χ÷–Ά®Ιΐ≤…”ΟΠΊΓΓ±ή‘–ΒΡ‘ΛΖά¥κ ©“‘ΩΊ÷ΤΖγœ’ΓΘ
‘ΛΖά¥κ ©Αϋά®Ϋχ––»―…ο ‘―ιΘ®»γ≤βΕ® HCG Π¬―«ΒΞΈΜΘ©ΓΔ≤…”ΟΗΏ–ßΒΡ±ή‘–ΖΫΖ®“‘ΦΑΫω‘Ύ»Ζ»œ‘¬Ψ≠ΤΎΚσ»κ―ΓΓΘ ‘―ιΤΎΦδΦλ≤β»―Θά…οΚΆΫ” ήΫΧ”ΐ”ΠΉψ“‘»Ζ±Θ‘Ύ“©Έο±©¬ΕΤΎΦδΘ®Ω…Ρή≥§Ιΐ ‘―ιΤΎœόΘ©Ήώ¥”ΈΣ±ή‘–Εχ…ηΦΤΒΡ¥κ ©ΓΘΈΣ÷ß≥÷’β–©ΖΫΖ®Θ§÷Σ«ιΆ§“β ι”ΠΑϋά®Υυ”–“―÷ΣΒΡ”κ…ζ÷≥ΕΨ–‘œύΙΊΒΡΠΈ–≈œΔΘ§»γ”κΫαΙΙΚΆ“©Γχάμ―ßΉς”ΟœύΙΊΒΡ“©Έο«±Γπ‘ΎΕΨ–‘ΒΡΉέΚœΤάΦέΓΘ»γΙϊΈ¥ΜώΒΟœύΙΊΒΡ…ζ÷≥ΕΨΓΦ–‘–≈œΔΘ§”ΠΥΒΟς«±‘ΎΒΡΖγœ’ΓΘ
‘ΎΥυ”–ICHΗςΖΫΘ§‘ΎΡ≥–©«ιΩωœ¬Θ§Έ¥Ϋχ––Ζ«ΝΌ¥≤…ζ÷≥ΕΨ–‘ ‘―ιΘ®»γ≈ΏΧΞ-ΧΞΉ– ‘―ιΘ©Θ§WOCBP“≤Ω…“‘»κ―ΓΝΌ¥≤ ‘―ιΓΘ“Μ÷÷«ιΩω «‘ΎΕΧΤΎΝΌ¥≤ ‘―ιΘ®»γ2÷ήΘ©÷–―œΟήΦύΩΊΜ≥‘–Ζγœ’ΓΘΝμ“Μ÷÷«ιΠγΩω «Θ§Β±Ηϋ≥ΛΗχ“©ΤΎœόΒΡΝΌ¥≤ ‘―ιΚΆ±©¬Ε®êΚœ ±Θ§ΗΟΦ≤≤Γ «≈°–‘®ëΗΏΖΔΘ®”≈ ΤΘ©Φ≤≤ΓΘ§≤ΜΡ…»κWOCBPΈόΖ®”–ΓΤΓΓ–ßΒΊ¥οΒΫΝΌ¥≤ ‘―ιΡΩΒΡΘ§≤Δ«“ΓβΕ‘Μ≥‘–Ζγœ’”–≥δΖ÷ΒΡΩΊ÷ΤΓΘ»±…ΌΖ«ΝΌ¥≤ ‘―ι…ζ÷≥ΕΨΠΖ –‘ ‘―ιΕχ‘ΎWOCBP…œΫχ––‘γΤΎ ‘―ιΒΡΤδΥϊΩΦ¬«“ρΥΊΘ§Αϋά®“©ΈοΒΡΉς”ΟΜζ÷Τ–≈œΔΓΔ“©Έοάύ–ΆΘ®»γΩΙΧεΘ©ΓΔΑκΥΞΤΎΓΔ“‘ΦΑΚœ ΒΡΕ·ΈοΡΘ–Ά…œΫχ––…ζ÷≥ΕΨ–‘ ‘―ιΒΡΡ―Ε»ΓΘ
Ά®≥ΘΘ§Β±ΜώΒΟΚœ ΠΖΒΡΝΫ÷÷Ε·Έο÷÷ τΒΡ≥θ≤Ϋ…ζ÷≥ΕΨ–‘Ή Νœ®êΘ®ΦϊΉΔ Ά3Θ©Θ§“‘ΦΑ≤…”ΟΝΥ≥δΖ÷ΒΡΓη±ή‘–¥κ ©Θ®ΦϊΉΔ Ά4Θ© ±Θ§‘ΎΆξ≥…Άξ’ϊΒΡ…ζ÷≥ΕΨ–‘ ‘Γθ―ι ±÷°«ΑΘ§Ω…‘Ύ“ΜœύΕ‘ΕΧΒΡΗχ“©ΤΎœόΡΎΘ®≤Μ≥§Ιΐ3Ηω‘¬Θ©Ϋ” ή“©ΈοΓό÷ΈΝΤ―–ΨΩ÷–Ρ…»κWOCBPΘ®÷ΝΕύ150»ΥΘ©ΓΘ’βΜυ”ΎΗΟ÷÷―υ±ΨΝΩΚΆΗχ“©ΤΎœόΒΡ”–ΩΊ÷ΤΝΌ¥≤ ‘―ιΓΐ÷–ΒΡΜ≥‘–Μζ¬ Ζ«≥ΘΒΆΘ®ΦϊΉΔ Ά5Θ©Θ§“‘ΦΑΈΣΦλ≤β¥σΕύ ΐ…ζ÷≥ΕΨ–‘”ΑœλΘ®ΤδΩ…“ΐΤπΝΌ¥≤ ‘―ι÷–Ρ…»κWOCBPΒΡΒΘ”«Θ©ΒΡ≥δΖ÷…ηΦΤ®}ΒΡ≥θ≤Ϋ ‘―ιΒΡΡήΝΠΓΘWOCBPΒΡ―υ±ΨΝΩΚΆ ‘Γϊ―ιΒΡΗχ“©ΤΎœό ήΒΫΩ…ΗΡΓΨ±δΜ≥‘–Μζ¬ ΒΡ»Υ»ΚΧΊ’ςΘ®»γΡξΝδΓΔΦ≤≤ΓΘ©ΒΡ”ΑœλΓΘ
‘ΎΟάΙζΘ§≈ΏΧΞΚΆΧΞΉ–ΖΔ”ΐΒΡΤάΦέΩ…Γϋ―”≥Ό÷Ν»κ―Γ”–Ω…Ρή…ζ”ΐ≤Δ≤…»Γ”––ß±ή‘–¥κ ©ΒΡΗΨ≈°ΒΡΔσΤΎΝΌ¥≤ ‘―ι«Α©•Άξ≥…ΓΘ‘Ύ≈ΖΟΥΚΆ»’±ΨΘ§≥ΐΝΥ…œ ωΥυΟη ωΒΡ«ιΩωΆβΘ§Άξ’ϊΒΡΖ«ΝΌ¥≤…ζ÷≥ΕΨ–‘ ‘―ι”Πœ»”ΎWOCBP±©¬Ε«ΑΆξ≥…ΓΘ
‘ΎΥυ”–ICHΗςΖΫΘ§ΈΣ÷ß≥÷ΔσΤΎΝΌ¥≤ ‘―ιΡ…ΓΗ»κWOCBPΘ§”ΠΆξ≥…ΧΊ“λ–‘ΒΡΩ…ΥΒΟς¥Τ–‘…ζ”ΐΝΠΒΡΖ«ΝΌ¥≤ ‘―ιΓΘ
‘ΎΥυ”–ICHΗςΖΫΘ§Έß≤ζΤΎ…ζ÷≥ΕΨ–‘ ‘―ι”Π‘Ύ…ξ«κ…œ – ±ΧαΫΜΘ§Μρ’ΏΘ§»γΙϊ”–“ΐΤπΒΘ”«ΒΡ‘≠“ρ ±”ΠΗϋ‘γΧαΫΜΓΘ
Β±Έ¥≤…”Ο”––ß±ή‘–¥κ ©ΒΡ”–Ω…Ρή…ζ”ΐΒΡΗΨ≈°Θ®ΦϊΉΔ Ά4Θ©Θ§Μρ»―…οΉ¥Ωω≤ΜΟςΒΡΗΨ≈°»κ―ΓΝΌ¥≤Γυ ‘―ι ±Θ§Υυ”–¥Τ–‘Ε·Έο…ζ÷≥ΕΨ–‘ ‘―ιΘ®≤ΈΩΦ10Θ©ΚΆ“≈¥ΪΕΨ–‘±ξΉΦΉιΚœ ‘―ιΘ®≤ΈΩΦ8Θ©Ψυ”Π‘ΎΝΌ¥≤ ‘―ιΩΣ Φ«ΑΆξ≥…ΓΘ
11.4 Μ≥‘–ΗΨ≈°
‘ΎΜ≥‘–ΗΨ≈°»κ―ΓΝΌ¥≤ ‘―ι«ΑΘ§”ΠΫχ––Ηςœν…ζ÷≥ΕΨ–‘ ‘Γΐ―ιΘ®≤ΈΩΦ10ΓΔ11Θ©ΚΆ“≈¥ΪΕΨ–‘±ξΉΦΉιΚœ ‘―ιΘ®≤ΈΩΦ8Θ©ΓΘΝμΆβΘ§“≤”ΠΤάΦέ“©Γ«Έο“‘ΆυΝΌ¥≤ Ι”ΟΒΡΑ≤»Ϊ–‘Ή ΝœΓΘ