发布日期2017-03-16 文 号:苏食药监械管〔2017〕45号
各设区市食品药品√监管局,昆山、泰兴、沭阳县(市)食品药品监管局,省局泰州医ζ药高新区直属分局,省∩医疗器械检验所,省局认证审评中心:
现将《江苏省医疗器※械生产企业重大事项报告程序》印发给你们,请认真◥贯彻实施。
江苏省食品药品监管局2017年3月16日江苏省医疗器械生产企业重大事项报告程序第一条 为做好医疗器械生产企业重大事项报告(以下简称“重大事项报告”)的管理工作,保证生产企业依法依规组织医疗器械生产,根据《医疗器械监督管理条例》及其配套规章、规范性文件的相关规定,结合我省实际,制订本程序。
第二条 本程序所称的重大↙事项指第一、二、三类医疗器械生产企业取得医疗器械生ぷ产备案凭证或医疗器械生产许可证后,其生产条件和质量管理体系运行相关设计开发、生产场所、设施/设备、生产环境、主要管理♂人员、生产工艺等发生重大变化、但依法不需要」进行医疗器械生产备案凭证或医疗器械生产许可证等备案/许可变更的事项。
本程序不包括医疗器械生产企业质量管理体系运行情况年度自查报告、医疗器械不良事件报告、医疗器械召回等法规已有规定程序的各类报告事项。
第三条 符合以下情形之一的,企业应当向省/市级医疗器械日常监管部★门(以下简称“省局”、“市局”)报告:(一)重大设计变更可能影∏响医疗器械安全、有效的;(二)核准的主要管理人员发生重大变化,可能对质量管理体系运行产生重大影响的;(三)已核准的生产厂房原址重¤建、布局重大调整、净化车间等功能区域同址改々建、扩建的;(四)关键工序、特殊过程、关键生产/检验设备、生产工艺等发生重大变化的;(五)医疗器械产品连续停产一年以①上且无同类产品◣在产的;(六)发生医疗器械重大生产质量事故的;(七)法规规定应当报告的其他情形。
第四条 符合下列情形之一的,不属于本程序所☉称的重大事项:(一)已注∴册的第二类、第三类医疗器械产品,产品名称、型号、规格、结构及ω组成、适用范围、产品技术要求、进口医疗器械生产□地址等发生变化,应当向原注册部门申请许可事项变更的;(二)已注◆册的第二类、第三∞类体外诊断试剂,产品名称、包装规格、主要组成成分、预期用途、产品技术要求、产品说明书、产▓品有效期、进口体外诊断试剂的生产地址等发生变化,应当向原注册部门申请许可事项变更或√首次注册的;(三)已注册的第二类、第三类医疗器械产品/体外诊断试剂,注册人名称和住所、代理人名称和住※所、境内生产地址等发生变化,应当向原注册部门申请登记事项变更的;(四)已备案的医疗器械/体外诊断试剂,备案信▲息表中登载内容及备案的产品技术要求发生变化的,备案人应当向原备案部门提出变更备案信」息的;(五)《医疗器械生产许可证》及其医疗器械生产产品登记『表载明的信息,如企业名称、法定代表人、企业负∑ 责人、住所、生产地址、生产范围、产品名称、注册号等发生变化,应当向原许可部门申请许可事项或登记事项变更的;(六)第ω 一类医疗器械/体外★诊断试剂生产备案凭证内容发生变化,应当变更备案的;(七)医疗器械不良事件报告及其年度报告;(八)医疗△器械质量管理体系年度自查报告;(九)医疗器械事中事后各类现场检查整改报告;(十)医疗器械∮召回相关报告。
第五条 重大事项报告遵循报告优先的原则,不能确认是否需要报告时,以①报告为第一选择。
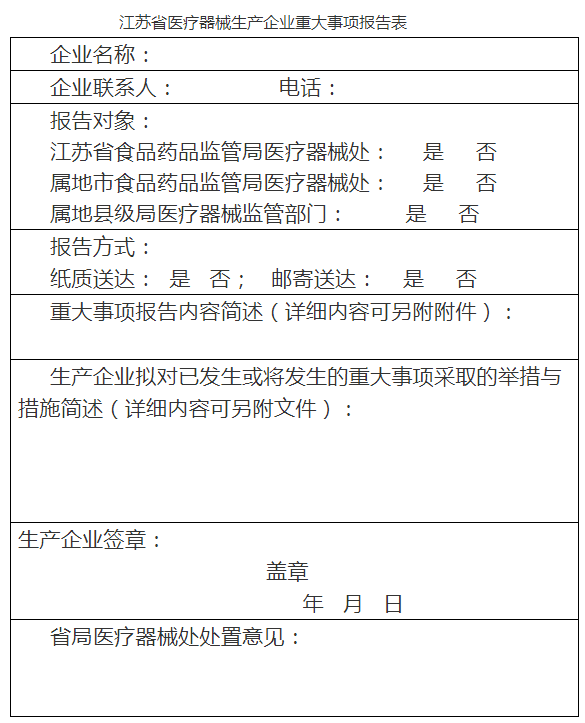
第六条 生产企业发生医疗器械相关法规或本程序规定应当报告的重大事项时,生产企业应当填写《江苏省医疗器械生产企业重大事项报告表》,同时向省局和市♀局报告。
第七条 省局、市局接到生产企业重大事项报告后,应当进行登记、沟通、分析,由省局依据法规和本程序提出处置意见并与市局共同实施。
第八条 省局提出的处置意见示例如下:(一)不符合重大事√项报告程序要求,生产企业应当∴根据相关法规规定进行医疗器械备案/注册,医疗器械生产备案/医疗器械生产许可;(二)不符合重大事项报告程序要求,生产企业应【当按相关法规规定履行义务(如可疑不良事件报告、医疗器械召回报告等);(三)符合重大事项报告程序要求,企业完成质量管理体系等变更后,由省局或市局』按照法规/规范要求进№行全部或部分项目现场检查或书面函审,并由市局组织监督整改※(必要时复查)。企业@ 完成整改后,由省局批准企业实施变更或按变更组织生∏产并通知市局,由市局记入企业监管档☆案。
(四)其他处置意见,如重大质量事故▼、重大舆情或群发事件等。
第九条 生产企业可以函寄重大事项报告▽书。省局、市局可通过约定的书面或电子信息方式传递企业重大事项报告相关信息。
第十条 仅涉及第一类医疗器〗械生产企业重大报告事项的,原则上由市局参照本程序全权︻处置,必要时报告省局。