来源:上海食药「监科技与信息
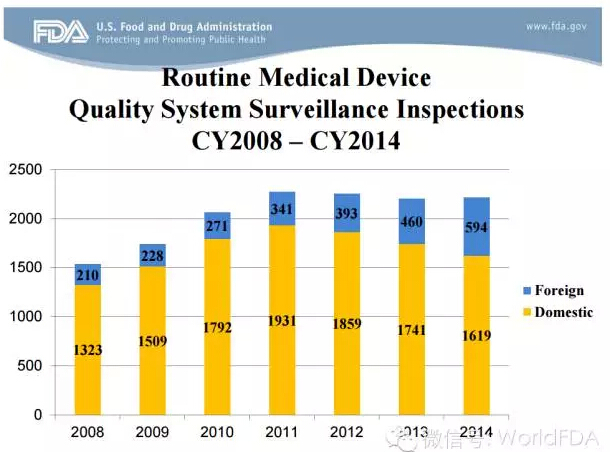
据FDA的器械和放射卫生中心(CDRH)的最新数据,2014年,FDA对器械产品的质量体系监督检查次数小幅增长,但是针对海外制造商的检查数量增加近30%。
按照海外制造商的国家来划分,对中国的检查占到海外检查的三成。
FDA此前已增加了海外仿制药生产商的检查数量,此数据表明ζ,在美国来自海外的产品迅速增加。

尽管质量⌒ 体系检查的数量有所增加,FDA发出的警告信数量在逐年减少,2012年160余个,2013年144个,到2014年的121个。超过60%的质量体系▓检查警告信是针对海外企业。
此外,在2014年,有3740个483表格提到了21 CFR 820(质量体系法规)不足。国内质量体系监督检查中,近一半∮的检查导致了483表格发出,而海外企业的相应检查中有57%的检查发出了483表格。
通过对警告信和483表格的检查情况分析,生产过程控制〓、纠正和预防措施仍然是最易发生违规的质量体系部分。
附:483表格和警告◥信的区别
483表格 | 警告信 | |
依据 | cGMP,不含21CFR211制剂条款 | 引用21CFR211制剂条款,原料药(API)除外 |
内容 | 现场检查所发现的不符合cGMP之处 | 最终讨论的违规项目 |
发出人 | 检查员 | FDA地区主任 |
发布形式 | 签发后向企业负责人当面宣读 | 签发后网站公布 |
回复要求 | 非强制性,但回复必↑须在15个工作日内 | 必须在15个工作日内回复,如不回复或回复不充分,会采取进一步法律制裁 |
纠正要求 | 自愿 | 强制 |
公开情况 | 依申请公开 | 主动公开,永久性 |
下一步制裁 | 警告信 | 拒@ 绝进口警报,停止∏药品申请审评 |